Basic Usage Tutorial#
cellpin imputes the missing genes of spatially-resolved cells by leveraging a single-cell RNA reference data.
This notebook walks through three complete workflows:
Section |
Description |
|---|---|
A — Standard |
Train on all overlapping genes and impute the full reference gene space |
B — Held-out evaluation |
Hold out panel genes before training to benchmark imputation accuracy |
Before running: update the file paths in Load data cell to point to your own .h5ad files.
Setup#
import cellpin
import torch
import scanpy as sc
import numpy as np
import yaml
from pathlib import Path
from time import time
Helper functions#
Two evaluation helpers used throughout the notebook:
correlation_panel— mean Pearson correlation between observed and imputed counts for the training panel genescorrelation_heldout— same metric, but restricted to genes that were held out during training (Option B only)
def correlation_panel(adata_imputed, layer_real, layer_imputed):
"""Mean per-gene Pearson r between two layers. Genes with -2 sentinel are excluded."""
counts_mat = adata_imputed.layers[layer_real]
imputed_mat = adata_imputed.layers[layer_imputed]
if hasattr(counts_mat, "toarray"):
counts_mat = counts_mat.toarray()
if hasattr(imputed_mat, "toarray"):
imputed_mat = imputed_mat.toarray()
valid_idx = np.where(~(counts_mat == -2).any(axis=0))[0]
pearsons = []
for j in valid_idx:
x, y = counts_mat[:, j], imputed_mat[:, j]
if np.std(x) > 0 and np.std(y) > 0:
pearsons.append(np.corrcoef(x, y)[0, 1])
else:
pearsons.append(np.nan)
mean_r = np.nanmean(pearsons)
print(f"Valid genes (no sentinel): {len(valid_idx)}")
print(f"Mean Pearson r : {mean_r:.4f}")
def correlation_heldout(adata_imputed, sp_adata_full, heldout_genes, layer_real="counts", layer_imputed="imputed"):
"""Evaluate imputation accuracy on genes held out during training."""
assert sp_adata_full.n_obs == adata_imputed.n_obs, "Cell count mismatch between sp_adata_full and adata_imputed."
pearsons = []
for gene in heldout_genes:
if gene not in sp_adata_full.var_names or gene not in adata_imputed.var_names:
print(f" [{gene}] not found — skipping")
continue
real = sp_adata_full[:, gene].layers[layer_real].toarray().flatten().astype(float)
imputed = adata_imputed[:, gene].layers[layer_imputed].toarray().flatten().astype(float)
if np.std(real) == 0 or np.std(imputed) == 0:
print(f" [{gene}] zero variance — skipping")
continue
r = np.corrcoef(real, imputed)[0, 1]
pearsons.append(r)
print(f" {gene:20s} Pearson = {r:.4f}")
mean_r = np.nanmean(pearsons)
print(f"\nMean Pearson over {len(pearsons)} held-out genes: {mean_r:.4f}")
A) Standard Workflow — Full Panel#
The standard cellpin workflow in four steps:
Load a single-cell reference (
sc_adata) and a spatial dataset (sp_adata)Run
cellpin.pp.setup_data()to align gene spacesTrain a
CellPinmodel on the scRNA referenceImpute the full gene space for every spatial cell
All overlapping genes between the two datasets are used as panel input.
A.1 — Load data#
Both objects need raw integer counts — either in .X or in a named layer (set LAYER accordingly).
sc_adata: single-cell reference atlas (full / highly-variable gene space)sp_adata: spatially-resolved dataset (panel genes only)
Example data
Source dataset: 10x Genomics Xenium breast preview dataset
First described in: Nature Communications (2023)
Processing: data were processed according to standard single-cell best practices
scRNA preprocessing: subset to Xenium panel genes plus 1,000 additional highly variable genes
Spatial data: subsampled, with a normalized layer added
Samples used:
Spatial: In Situ Sample 1, Replicate 1
Single-cell: FRP
LAYER = "counts" # layer key with raw integer counts; set to None to use .X
sc_adata = cellpin.pp.load_sc_example()
sp_adata = cellpin.pp.load_sp_example()
print(f"sc_adata : {sc_adata.n_obs:,} cells × {sc_adata.n_vars:,} genes")
print(f"sp_adata : {sp_adata.n_obs:,} cells × {sp_adata.n_vars:,} spatial genes")
sc_adata : 23,867 cells × 1,307 genes
sp_adata : 30,600 cells × 313 spatial genes
A.2 — Gene alignment check#
Confirm which spatial genes are present in the scRNA reference before calling setup_data().
Any missing genes will be dropped automatically, but it is good to know upfront.
sc_genes = sc_adata.var_names
sp_genes = sp_adata.var_names
missing = sp_genes.difference(sc_genes)
if len(missing) == 0:
print(f"All {len(sp_genes)} spatial genes are present in sc_adata ✓")
else:
print(f"WARNING: {len(missing)} spatial gene(s) not in sc_adata (will be dropped by setup):")
print(sorted(missing))
overlap = sp_genes.intersection(sc_genes)
print(f"\nPanel size (overlap) : {len(overlap)} genes")
print(f"scRNA-only genes (to impute): {len(sc_genes) - len(overlap)} genes")
WARNING: 6 spatial gene(s) not in sc_adata (will be dropped by setup):
['AKR1C1', 'ANGPT2', 'BTNL9', 'CD8B', 'POLR2J3', 'TPSAB1']
Panel size (overlap) : 307 genes
scRNA-only genes (to impute): 1000 genes
A.3 — Setup and train#
sc_dataset, sp_dataset = cellpin.pp.setup_data(
sc_adata, sp_adata, layer=LAYER
) # add batch_key="batch" if needed -recommended for reference atlases
============================================================
[cellpin.pp.setup] Setting up CellPin datasets
============================================================
[cellpin.pp.setup] sc_adata : 23,867 cells × 1,307 genes
[cellpin.pp.setup] st_adata : 30,600 cells × 313 spatial genes
[cellpin.pp.setup] Expression read from layer='counts'
[cellpin.pp.setup] WARNING: 6 spatial gene(s) not found in sc_adata — will be dropped:
['AKR1C1', 'ANGPT2', 'BTNL9', 'CD8B', 'POLR2J3', 'TPSAB1']
[cellpin.pp.setup] Panel : 307 genes overlap (98.1% of spatial genes retained)
[cellpin.pp.setup] Imputed : 1,307 genes total in sc space (1,000 genes to impute, not in panel)
[cellpin.pp.setup] sc_dataset: 23,867 cells, 1,307 genes total, 307 panel genes
[cellpin.pp.setup] st_dataset: 30,600 cells, 307 panel genes
time_start = time()
model_A = cellpin.CellPin(sc_dataset)
model_A.fit(
sc_dataset, pretrain_epochs=50, train_epochs=60, save_checkpoints=False
) # save_checkpoints=True is much slower and not needed for this tutorial
print(f"Training time: {(time() - time_start) / 60:.2f} minutes")
Epoch 49: 100%|██████████| 75/75 [00:02<00:00, 25.20it/s, v_num=0, val_loss=457.0, val_reconst_loss=438.0, val_kl_loss=16.60, val_kl_l_loss=2.350, val_pearson_loss=0.484, train_loss=465.0, train_reconst_loss=445.0, train_kl_loss=17.40, train_kl_l_loss=2.390, train_pearson_loss=0.464]
Epoch 58: 100%|██████████| 75/75 [00:03<00:00, 20.83it/s, v_num=0, val_loss=524.0, val_reconst_loss=438.0, val_kl_loss=55.80, val_kl_l_loss=2.490, val_distill_loss=0.0722, val_snn_loss=1.630, val_inv_loss=0.210, val_snn_temperature=0.104, val_pearson_loss=0.468, train_loss=520.0, train_reconst_loss=435.0, train_kl_loss=59.30, train_kl_l_loss=2.510, train_distill_loss=0.0816, train_snn_loss=1.670, train_inv_loss=0.223, train_snn_temperature=0.104, train_pearson_loss=0.436]
Training time: 6.10 minutes
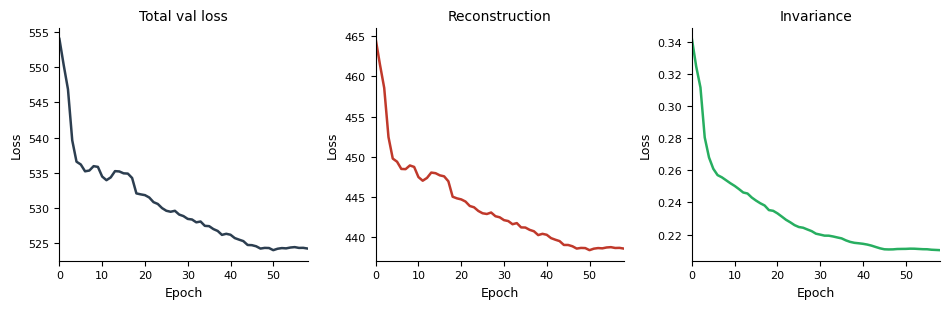
model_A.pl.losses(smooth=5)
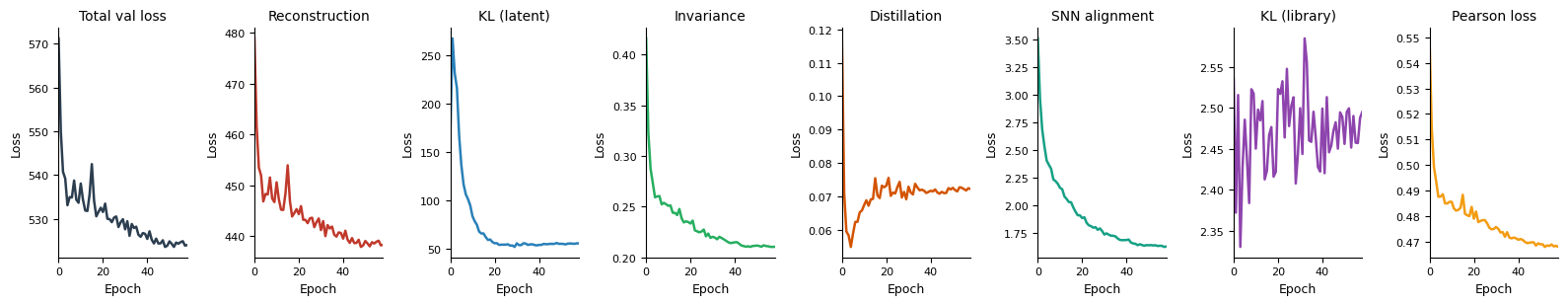
model_A.pl.losses(keys="all")
A.4 — Impute#
return_norm=True adds an area-normalised log1p layer (imputed_norm).
Set area_key=None if your spatial data has no cell_area column.
time_start = time()
dl_A = torch.utils.data.DataLoader(sp_dataset, batch_size=512, shuffle=False)
adata_imputed_A = model_A.impute(
dl_A,
obs_adata=sp_adata,
return_norm=True,
area_key="cell_area",
nb_count_samples=20,
#return_int=True is recomended for denoising data and performing downstream analyses
)
print(f"Imputation time: {(time() - time_start) / 60:.2f} minutes")
print(adata_imputed_A)
Embedding and imputing cells (MC, 50 samples)...
[CellPin.impute] Panel gene order confirmed ✓
[impute] Filling 1000 gene(s) absent from obs_adata layers with sentinel -2.0
[impute] Filling 1000 gene(s) absent from obs_adata layers with sentinel -2.0
Imputation time: 1.15 minutes
AnnData object with n_obs × n_vars = 30600 × 1307
obs: 'cell_id', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'region'
obsm: 'X_cellpin', 'spatial'
layers: 'counts', 'log1p_norm', 'imputed', 'imputed_norm'
A.5 — Evaluate#
Pearson correlation between observed and imputed counts for panel genes.
print("=== Count space ===")
correlation_panel(adata_imputed_A, layer_real="counts", layer_imputed="imputed")
print("\n=== Log-normalised space ===")
correlation_panel(adata_imputed_A, layer_real="log1p_norm", layer_imputed="imputed_norm")
=== Count space ===
Valid genes (no sentinel): 307
Mean Pearson r : 0.5938
=== Log-normalised space ===
Valid genes (no sentinel): 307
Mean Pearson r : 0.4980
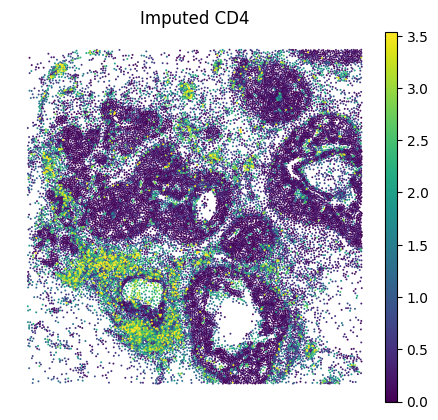
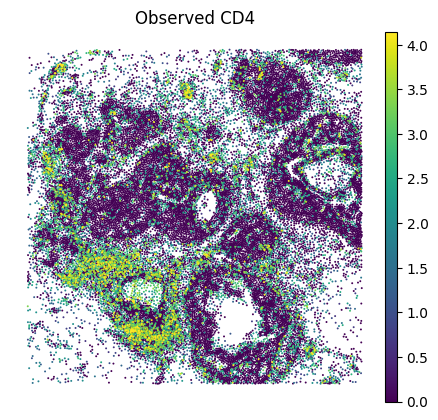
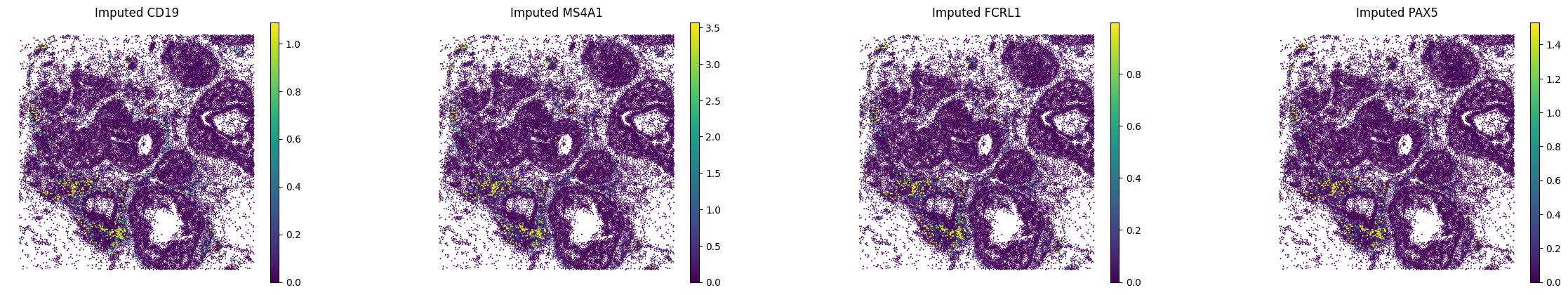
A.6 — Visualise#
Spatial maps and UMAP for a few marker genes.
MS4A1 and CD19 were in the panel; FCRL1 and PAX5 are purely imputed. (All B cell genes, expected to co-localise)
GENE = "CD4" # edit as needed
sc.pl.spatial(
adata_imputed_A, layer="imputed_norm", color=GENE, spot_size=15, vmax="p99", frameon=False, title=f"Imputed {GENE}"
)
sc.pl.spatial(
adata_imputed_A, layer="log1p_norm", color=GENE, spot_size=15, vmax="p99", frameon=False, title=f"Observed {GENE}"
)
bcells = ["CD19", "MS4A1", "FCRL1", "PAX5"]
print("Spatial plots — B-cell markers:")
sc.pl.spatial(
adata_imputed_A,
layer="imputed_norm",
color=bcells,
spot_size=15,
vmax="p99",
frameon=False,
title=[f"Imputed {g}" for g in bcells],
)
sc.pl.spatial(
adata_imputed_A,
layer="log1p_norm",
color=bcells,
spot_size=15,
vmax="p99",
frameon=False,
title=[f"Observed {g}" for g in bcells],
)
Spatial plots — B-cell markers:
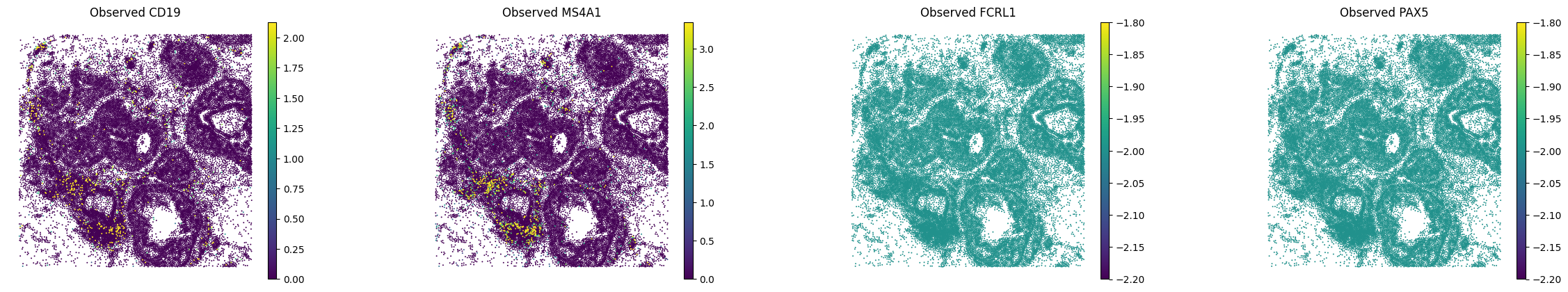
A.7 — UMAP from cellpin embeddings#
Cell embeddings in obsm["X_cellpin"] can be used directly to build a neighborhood graph and compute a UMAP
sc.pp.neighbors(adata_imputed_A, use_rep="X_cellpin")
sc.tl.umap(adata_imputed_A)
print("UMAP — B-cell markers:")
sc.pl.umap(adata_imputed_A, color=bcells, layer="imputed_norm", vmax="p99", title=[f"Imputed {g}" for g in bcells],frameon=False)
sc.pl.umap(adata_imputed_A, color=bcells, layer="log1p_norm", vmax="p99", title=[f"Observed {g}" for g in bcells],frameon=False)
UMAP — B-cell markers:
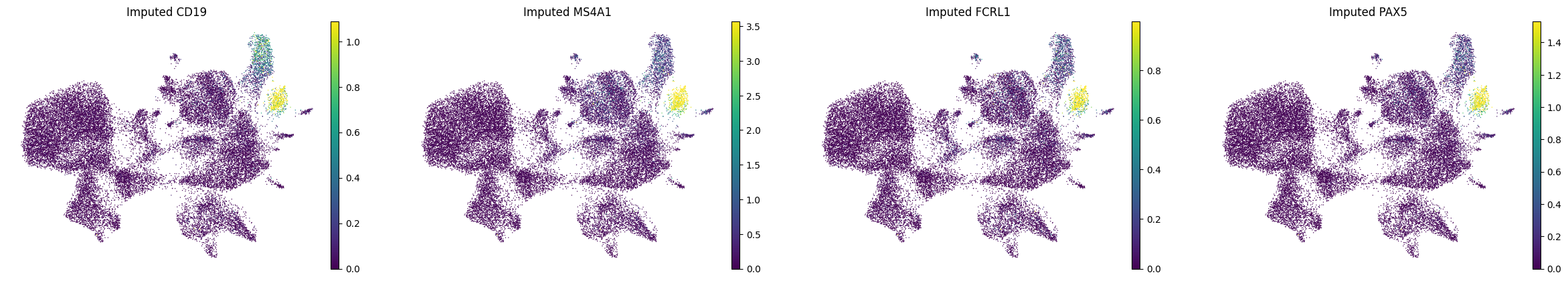
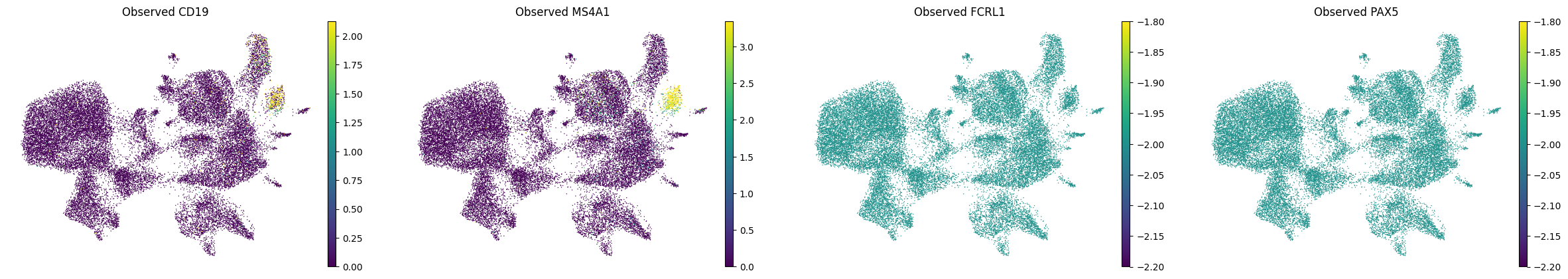
B) Held-Out Gene Evaluation#
To benchmark how well cellpin imputes genes it has never seen during training, we:
Randomly sample a fraction of panel genes as held-out genes
Remove them from the spatial input before
setup_data()Train and impute as normal
Compare imputed values against the ground-truth counts for those held-out genes
The held-out Pearson correlation is the most honest imputation benchmark.
B.1 — Define held-out genes#
rng = np.random.default_rng(42)
# Panel genes in sc_adata order — consistent with setup_data()'s internal ordering
panel_genes_all = [g for g in sc_adata.var_names if g in set(sp_adata.var_names)]
print(f"Full panel size: {len(panel_genes_all)} genes")
HOLDOUT_FRACTION = 0.10 # hold out 10 % of the panel
n_holdout = max(1, int(len(panel_genes_all) * HOLDOUT_FRACTION))
heldout_genes = rng.choice(panel_genes_all, size=n_holdout, replace=False).tolist()
training_genes = [g for g in panel_genes_all if g not in heldout_genes]
print(f"Held-out genes : {n_holdout} → {heldout_genes[:5]} ...")
print(f"Training panel : {len(training_genes)} genes")
Full panel size: 307 genes
Held-out genes : 30 → ['NDUFA4L2', 'CCDC6', 'LEP', 'CCL8', 'ELF3'] ...
Training panel : 277 genes
# Build the reduced spatial object (held-out genes removed)
sp_adata_reduced = sp_adata[:, training_genes].copy()
assert len(set(heldout_genes) & set(sp_adata_reduced.var_names)) == 0, (
"Held-out genes still present in reduced sp_adata!"
)
print(
f"sp_adata_reduced: {sp_adata_reduced.n_obs:,} cells × {sp_adata_reduced.n_vars:,} genes (held-out genes removed ✓)"
)
sp_adata_reduced: 30,600 cells × 277 genes (held-out genes removed ✓)
B.2 — Setup and train#
sc_dataset_B, sp_dataset_B = cellpin.pp.setup_data(sc_adata, sp_adata_reduced, layer=LAYER)
============================================================
[cellpin.pp.setup] Setting up CellPin datasets
============================================================
[cellpin.pp.setup] sc_adata : 23,867 cells × 1,307 genes
[cellpin.pp.setup] st_adata : 30,600 cells × 277 spatial genes
[cellpin.pp.setup] Expression read from layer='counts'
[cellpin.pp.setup] Panel : 277 genes overlap (100.0% of spatial genes retained)
[cellpin.pp.setup] Imputed : 1,307 genes total in sc space (1,030 genes to impute, not in panel)
[cellpin.pp.setup] sc_dataset: 23,867 cells, 1,307 genes total, 277 panel genes
[cellpin.pp.setup] st_dataset: 30,600 cells, 277 panel genes
model_B = cellpin.CellPin(sc_dataset_B)
model_B.fit(
sc_dataset_B, pretrain_epochs=50, train_epochs=60, save_checkpoints=False
) # save_checkpoints=True is much slower and not needed for this tutorial
Epoch 0: 39%|███▊ | 29/75 [00:01<00:01, 25.61it/s, v_num=1]
Epoch 49: 100%|██████████| 75/75 [00:03<00:00, 23.22it/s, v_num=1, val_loss=456.0, val_reconst_loss=436.0, val_kl_loss=17.00, val_kl_l_loss=2.390, val_pearson_loss=0.483, train_loss=464.0, train_reconst_loss=444.0, train_kl_loss=17.70, train_kl_l_loss=2.380, train_pearson_loss=0.465]
Epoch 59: 100%|██████████| 75/75 [00:03<00:00, 19.93it/s, v_num=1, val_loss=521.0, val_reconst_loss=436.0, val_kl_loss=55.60, val_kl_l_loss=2.470, val_distill_loss=0.0726, val_snn_loss=1.660, val_inv_loss=0.214, val_snn_temperature=0.104, val_pearson_loss=0.470, train_loss=522.0, train_reconst_loss=436.0, train_kl_loss=59.50, train_kl_l_loss=2.520, train_distill_loss=0.0836, train_snn_loss=1.730, train_inv_loss=0.231, train_snn_temperature=0.104, train_pearson_loss=0.440]
B.3 — Impute#
dl_B = torch.utils.data.DataLoader(sp_dataset_B, batch_size=512, shuffle=False)
adata_imputed_B = model_B.impute(
dl_B,
obs_adata=sp_adata_reduced,
return_norm=True,
area_key="cell_area",
nb_count_samples=20,
#return_int=True is recomended for denoising data and performing downstream analyses
)
print(adata_imputed_B)
Embedding and imputing cells (MC, 50 samples)...
[CellPin.impute] Panel gene order confirmed ✓
[impute] Filling 1030 gene(s) absent from obs_adata layers with sentinel -2.0
[impute] Filling 1030 gene(s) absent from obs_adata layers with sentinel -2.0
AnnData object with n_obs × n_vars = 30600 × 1307
obs: 'cell_id', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'region'
obsm: 'X_cellpin', 'spatial'
layers: 'counts', 'log1p_norm', 'imputed', 'imputed_norm'
B.4 — Evaluate#
Two metrics:
Panel Pearson — reconstruction quality on the training genes (sanity check)
Held-out Pearson — true imputation benchmark on genes the model never saw
print("=== Training panel genes (count space) ===")
correlation_panel(adata_imputed_B, layer_real="counts", layer_imputed="imputed")
=== Training panel genes (count space) ===
Valid genes (no sentinel): 277
Mean Pearson r : 0.5963
print("=== Held-out genes — count space ===")
correlation_heldout(
adata_imputed_B,
sp_adata_full=sp_adata, # sp_adata (full) has ground-truth counts for held-out genes
heldout_genes=heldout_genes,
layer_real="counts",
layer_imputed="imputed",
)
=== Held-out genes — count space ===
NDUFA4L2 Pearson = 0.1975
CCDC6 Pearson = 0.6173
LEP Pearson = 0.3429
CCL8 Pearson = 0.1985
ELF3 Pearson = 0.6551
ENAH Pearson = 0.4506
TIMP4 Pearson = 0.2919
KRT14 Pearson = 0.8070
ITGAX Pearson = 0.7537
LYZ Pearson = 0.7676
GNLY Pearson = 0.5750
CX3CR1 Pearson = 0.4900
SPIB Pearson = 0.3898
CXCL12 Pearson = 0.8441
KRT23 Pearson = 0.8277
DNAAF1 Pearson = 0.0584
AGR3 Pearson = 0.6253
FOXA1 Pearson = 0.8592
FOXP3 Pearson = 0.3508
PTN Pearson = 0.7818
PDCD1LG2 Pearson = 0.2287
CCND1 Pearson = 0.7912
SQLE Pearson = 0.4533
ANKRD30A Pearson = 0.8562
SERPINA3 Pearson = 0.7612
CEACAM8 Pearson = -0.0366
GPR183 Pearson = 0.5304
CLECL1 Pearson = 0.2589
LILRA4 Pearson = 0.7407
CCPG1 Pearson = 0.4594
Mean Pearson over 30 held-out genes: 0.5309
print("=== Held-out genes — log-normalised space ===")
correlation_heldout(
adata_imputed_B,
sp_adata_full=sp_adata,
heldout_genes=heldout_genes,
layer_real="log1p_norm",
layer_imputed="imputed_norm",
)
=== Held-out genes — log-normalised space ===
NDUFA4L2 Pearson = 0.1717
CCDC6 Pearson = 0.5261
LEP Pearson = 0.1451
CCL8 Pearson = 0.1529
ELF3 Pearson = 0.6506
ENAH Pearson = 0.3184
TIMP4 Pearson = 0.0974
KRT14 Pearson = 0.7578
ITGAX Pearson = 0.5899
LYZ Pearson = 0.6539
GNLY Pearson = 0.3986
CX3CR1 Pearson = 0.4187
SPIB Pearson = 0.3675
CXCL12 Pearson = 0.7626
KRT23 Pearson = 0.7281
DNAAF1 Pearson = 0.0097
AGR3 Pearson = 0.5790
FOXA1 Pearson = 0.8434
FOXP3 Pearson = 0.3118
PTN Pearson = 0.7032
PDCD1LG2 Pearson = 0.1542
CCND1 Pearson = 0.6785
SQLE Pearson = 0.3021
ANKRD30A Pearson = 0.8321
SERPINA3 Pearson = 0.6422
CEACAM8 Pearson = -0.0062
GPR183 Pearson = 0.5405
CLECL1 Pearson = 0.2273
LILRA4 Pearson = 0.5649
CCPG1 Pearson = 0.2203
Mean Pearson over 30 held-out genes: 0.4448
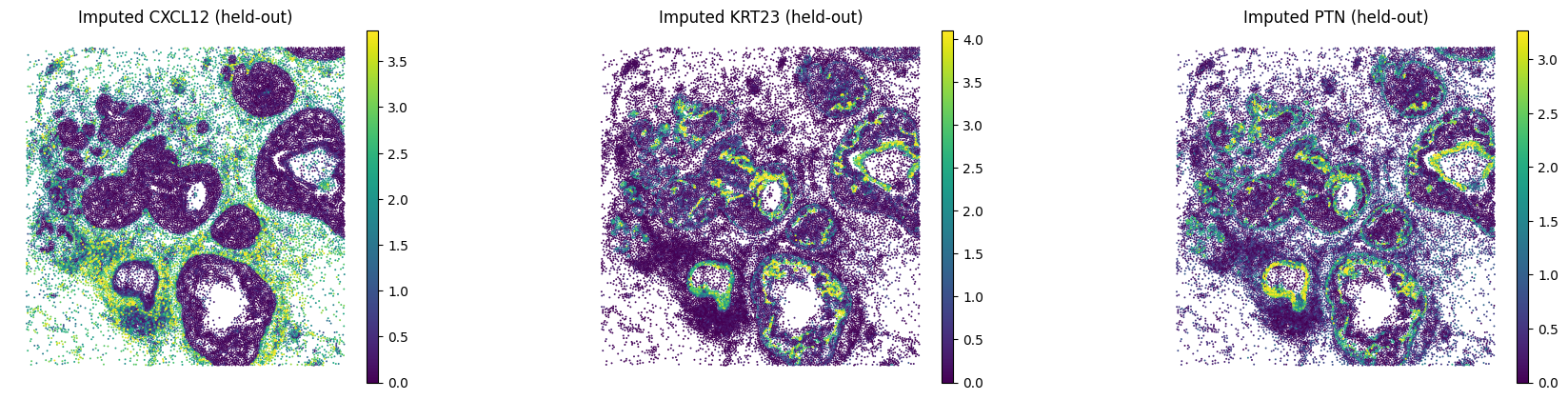
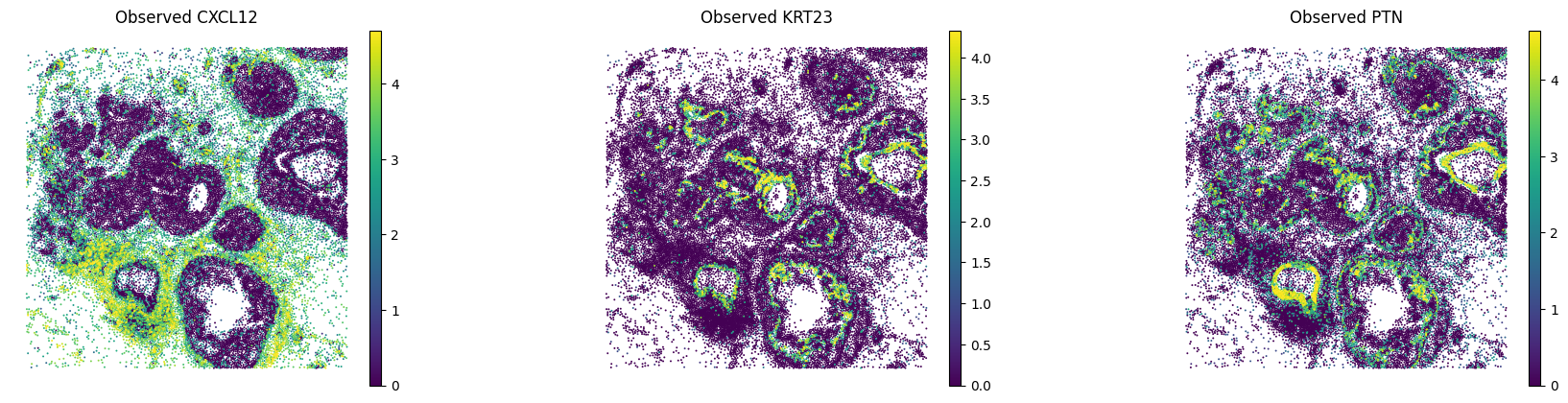
B.5 — Visualise held-out genes#
Compare imputed vs. observed spatial expression for a few held-out genes with strong Pearson correlation.
# Edit: pick held-out genes with high Pearson from the evaluation above
GENES_HELDOUT = ["CXCL12", "KRT23", "PTN"]
sc.pl.spatial(
adata_imputed_B,
layer="imputed_norm",
color=GENES_HELDOUT,
spot_size=15,
vmax="p99",
frameon=False,
title=[f"Imputed {g} (held-out)" for g in GENES_HELDOUT],
)
sc.pl.spatial(
sp_adata,
layer="log1p_norm",
color=GENES_HELDOUT,
spot_size=15,
vmax="p99",
frameon=False,
title=[f"Observed {g}" for g in GENES_HELDOUT],
)